Vaskulitis: Arten (kutane, Purpura, leukozytoklastische, urticarial)
Die Vaskulitis (oder Angiitis) ist eine Entzündung der Blutgefäße (Arterien, Venen und Kapillaren), die von Nekrosen (Zelltod) betroffen sein kann.
Es handelt sich um eine seltene Pathologie, die Menschen jeden Alters betreffen kann.
Diese systemische Erkrankung betrifft das Gefäßsystem und verändert die primäre Funktion:
- Der Arterien, die sauerstoffreiches Blut zu den Körpergeweben transportieren,
- Der Venen, die sauerstoffarmes Blut zum Herzen zurückführen.
Die Vaskulitis ist eine Multisystemerkrankung, die folgende Organe betreffen kann:
- Haut,
- Nieren,
- Herz,
- Nerven,
- Lungen,
- Gelenke,
- Darm,
- Augen,
- Ohren,
- Nase.
Inhalt
Klassifikation der Vaskulitis
Vaskulitiden unterteilen sich in:
- Primäre – idiopathische, das bedeutet, man kennt die Ursache nicht,
- Sekundäre – sie werden verursacht durch: Infektionskrankheiten, Krankheiten des Bindegewebes, Tumore oder Kryoglobulinämie, Einnahme von Medikamenten, Bestrahlung, chemische Wirkstoffe, Organabstoßung usw.
Klassifikation nach Größe der betroffenen Blutgefäße
2011-2012 übernommene Bezeichnungen durch International Chapel Hill Consensus Conference Nomenclature of the Vasculitides
Nach Blutgefäßen Groß 1. Takayasu-Arteriitis
- Riesenzellarteriitis Mittelgroß 1. Polyarteriitis nodosa
- Kawasaki-Krankheit Klein 1. ANCA-assoziierte Vaskulitiden
– Mikroskopische Polyangiitis
– Wegener-Granulomatose
– Eosinophile Granulomatose mit eosinophiler Polyangiitis 2. Immunkomplex-Vaskulitis
– Goodpasture-Syndrom
– Kryoglobulinämische Vaskulitis
– Systemische IgA-Vaskulitis
– Hypokomplementämische urtikarielle Vaskulitis Variabel 1. Behcet-Krankheit
- Cogan-Syndrom
Andere Arten Einzelnes
Organ
- Kutane leukozytoklastische Vaskulitis
- Kutane Arteriitis
- Primäre Vaskulitis des Zentralnervensystems
- Isolierte Aortitis
- Sonstige Systemische
Erkrankungen
- Vaskulitis bei Lupus
- Vaskulitis bei rheumatoider Arthritis
- Sarkoidose-Vaskulitis
- Sonstige
Mögliche Ursache
- Kryoglobulinämische Vaskulitis bei Virus-C-Hepatitis
- Vaskulitis bei Virus-B-Hepatitis
- Aortitis bei Syphilis
- Immunkomplexvaskulitis im Zusammenhang mit Medikamenten
- Vaskulitis durch ANCA-Medikamente
- Vaskulitis im Zusammenhang mit Krebs
- Sonstige
Beschreibung der Vaskulitisarten
Vaskulitis der großen Gefäße
- Riesenzellarteriitis (oder Temporalis);
- Takayasu-Arteriitis;
Die Takayasu-Arteriitis und die Arteriitis temporalis (bei Erwachsenen) treten an den großen elastischen Arterien auf und zeigen ähnliche histologische Merkmale (des Gewebes).
Dabei ergeben sich folgende Schritte:
- Dendritische Zellen in der Tunica adventitia (äußere Schicht) der Arterie begünstigen und stimulieren CD4+-Zellen.
- Diese Zellen aktivieren Monozyten und Makrophagen, die Verletzungen der Gefäßwand verursachen.
Gefäßläsionen sind durch eine Entzündung mit Eindringen in Lymphozyten und Monozyten in allen Arterienwandschichten gekennzeichnet.
In der Regel bilden aktivierte T-Zellen und Makrophagen Granulome, in denen auch mehrkernige Riesenzellen vorhanden sind.
Oft kommt es zur Verengung des Blutgefäßes, weil in der inneren Tunica eine wichtige Zellproliferation und damit eine Volumenzunahme vorliegt.
Die Takayasu-Arteriitis betrifft vorwiegend die Brustaorta in Höhe des Aortenbogens und seine großen Nebenzweige, besonders die Arteria subclavia.
In 30 % der Fälle sind betroffen:
- Andere Abschnitte der Aorta,
- Nierenarterien,
- Koronararterien (die Beteiligung der Koronararterien kann zu relevanten kardiologischen Problemen führen, insbesondere zu Herzinfarkt und Tod).
In 50 % der Fälle können auch die Lungenarterien betroffen sein.
Man kennt den Grund dafür nicht, doch Wissenschaftler nehmen an, dass die Ursache sein könnte:
- Infektionen mit Treponema pallidum, Streptokokken und das Mykobakterium der Tuberkulose,
- Geschwächtes Immunsystem (aufgrund von Autoimmunerkrankungen, Morbus Crohn, Colitis ulcerosa, Sarkoidose usw.)
In der Aorta kommt es zu einer Verengung des Gefäßlumens, die zum Verschwinden des arteriellen Pulses führt (daher wird die Erkrankung als Pulslos-Krankheit oder Aortenbogensyndrom bezeichnet).
Die Entwicklung der Krankheit kann schnell erfolgen, sie kann eine Dauer von ein bis zwei Jahren haben.
Nachdem die Symptome vergangen sind und die Überlebensrate gut ist, können einige neurologische und visuelle Störungen bestehen bleiben.

Vaskulitis der mittelgroßen Gefäße
Dieser Typ der Vaskulitis betrifft vor allem die mittleren Arterien, das sind vor allem die viszeralen Arterien und ihre Verzweigungen.
Es treten Entzündungen der Muskelarterien auf.
Die Fibrinoidnekrose ist typisch für die mittelgroßen Gefäße und kann zur Bildung führen von:
- Knoten (Gefäßverengung),
- Aneurysma.
Mögliche Ursachen im Falle der Kawasaki-Krankheit sind:
- Infektionen,
- Aktivierung der Lymphozyten.
Die auslösenden Faktoren der Polyarteriitis nodosa sind unklar, können aber durch Hepatitis B oder C verursacht sein.
Die Kawasaki-Krankheit (kann Rhythmusstörungen, Perikarditis, Hautausschläge und Entzündungen im Mund hervorrufen);
Die Ursache ist unbekannt, doch die zuverlässigste Hypothese ist die Parvovirus-B19-Infektion, die die Zellen des Lymphatischen Systems und des Blutes (rote Blutkörperchen, Erythroblasten usw.) betrifft.
Andere Hypothesen betrachten:
- Toxische Substanzen,
- Allergene.
Oftmals sind die Herzkranzgefäße betroffen (wie bei der Polyarteriitis nodosa), damit kann eine Differenzialdiagnose schwierig sein.
Die Entwicklung von Koronararterienläsionen kann zur Bildung von Aneurysmen führen.
In den meisten Fällen ist die Prognose gut, doch in 1-3 % der Fälle kann durch die Vaskulitis der plötzliche Tod eintreten durch:
- Platzen von Aneurysmen der Herzkranzgefäße,
- Thrombose der Herzkranzarterien,
- Infarkt,
- Herzmuskelentzündung.
Diese Erkrankung kann jedes Organ betreffen (Niere, Herz, Leber, Darm, Hoden usw.).
Es können viele Symptome auftreten und:
- Unspezifisch sein,
- Systemisch sein.
Vor allem hat der Patient Fieber und einen unerklärlichen Gewichtsverlust.
Oftmals leidet der Patient auch unter:
- Bluthochdruck,
- Muskelschmerzen,
- Durchfall,
- Übelkeit und Erbrechen,
- Nervenentzündung,
- Meläna (verdautes Blut im Stuhl) aufgrund von Darmgeschwüren,
- Bauchschmerzen.
Der Verlauf ist unterschiedlich, er kann chronisch oder intermittierend sein sowie mit Perioden langer Remissionsphasen einhergehen.
Man beobachtet eine nekrotisierende Entzündung, die die Arterienwand fokal (lokalisiert) befällt und/oder den gesamten Gefäßumfang betrifft.
Im Laufe der Zeit bildet sich eine Fibrose.
Die bedeutendste Komplikation ist der renale Hochdruck.
In typischen Fällen zeigt die Angiographie das Vorliegen von Aneurysmen in den viszeralen Arterien.
Wie lange dauert die Erkrankung? Die Prognose bleibt schwerwiegend, trotz der Therapie mit Immunsuppressiva.
Vaskulitis der kleinen Gefäße
Durch Antikörper vermittelte Vaskulitis
ANCA-Antikörper (Anti-Neutrophile cytoplasmatische Antikörper) sind auf Proteine gerichtet, die sich im Zytoplasma von Neutrophilen und Monozyten (zum Beispiel PR3, MPO) und auch auf den Zelloberflächen befinden.
IgG ANCA können direkt aktivieren:
- Die Neutrophilen,
- Die Monozyten.
Diese aktivierten Zellen reagieren mit den Zellen des Endothels durch Adhäsionsmoleküle, das sind Rezeptoren (Proteine) auf der Zellmembran.
Sie setzen außerdem Entzündungsmediatoren frei (Substanzen, die Entzündungen verursachen) wie Granzyme (Enzyme, die chemische Verbindungen in Proteine spalten) und reaktive Sauerstoffmetabolite.
Die Folgen sind:
- Apoptosis,
- Nekrose.
Außerdem kann das Anti-MPO-IgG das MPO aktivieren, das schwere endotheliale Schäden verursacht.
Die aktivierten ANCA-Neutrophile können Faktoren freisetzen, die den alternativen Weg des Komplements (Körperabwehrsystem) aktivieren.
Auf diese Weise beginnt ein Zyklus der Amplifikation (zellulärer Vorgang), der die nekrotisierende Entzündung verursacht. (Jennette JC, Falk RJ. Neue Einblicke in die Pathogenese der Vaskulitis im Zusammenhang mit antineutrophilen zytoplasmatischen Autoantikörpern. Curr Opin Rheumatol. 2008 Jan. 20(1): 55-60)
Antikörperablagerung
Die Purpura Schoenlein-Henoch ist im Allgemeinen durch die Ablagerung von IgA-Antikörpern in den betroffenen Geweben gekennzeichnet.
Die Hautbiopsie zeigt eine leukozytoklastische Vaskulitis mit Akkumulation an, und zwar der:
- Neutrophilen,
- Lymphozyten,
- Monozyten.
Zusätzlich werden auch IgA, C3 und Fibrin vorgefunden:
- In den Wänden der betroffenen Blutgefäße und in den postkapillären Venolen in der Dermis,
- In den endothelialen und mesangialen Zellen der Niere.
Bei manchen Patienten kann ein erhöhtes IgA im Blut vorliegen.
Die Purpura Schoenlein-Henoch befällt vor allem Kinder.
Die Vaskulitis der kleinen Arterien wird unterteilt in:
- ANCA positiv: wenn Antikörper gegen zytoplasmatische Antigene der Neutrophilen beteiligt sind
- ANCA-negativ: in anderen Fällen.
Beim Goodpasture-Syndrom binden sich die zirkulierenden Antikörper an das Typ-IV-Kollagen im Inneren der Basalmembran des Nierenkörperchens.
Lungenblutungen treten auf, wenn diese Antikörper die Basalmembran der Alveolen durchdringen.
Das tritt oft mit einer nekrotisierenden Glomerulonephritis auf, die eine signifikante Nierenfunktionsstörung verursachen kann. (Ozen S, Anton J, Arisoy N, et al. Juvenile polyarteritis: results of a multicenter survey of 110 children. J Pediatr. 2004 Oct. 145(4): 517-22).
Die mikroskopische Polyangiitis (mPA) ist eine nekrotisierende Vaskulitis, gekennzeichnet durch:
- Glomerulonephritis,
- Lungenkapillarentzündung.
Zu den Symptomen gehören:
- Livider Hautausschlag,
- Eiweiß im Urin,
- Blut im Urin,
- Blutig tingierter Husten,
- ZNS-Dysfunktion,
- Gelenkschmerzen.
Beim Churg-Strauss-Syndrom und der Granulomatose mit Polyangiitis bilden sich um und außerhalb der Gefäße Granulome.
Bei den anderen Vaskulitiden treten Entzündungen nur in den kleinen Gefäßen auf.
Im Gewebebereich basiert die Diagnose auf drei Parametern:
- Penetration der Eosinophilen,
- Nekrotisierende Vaskulitis,
- Perivaskuläre und extravaskuläre Granulome mit Bildung von Fibrose und Verkalkungen.
Jedoch tritt dies nie gleichzeitig auf.
Die Granulomatose mit Polyangiitis (zum Beispiel die Wegener-Granulomatose) betrifft vor allem das Atmungssystem, kann aber auch jedes andere Organ betreffen.
Charakteristisch können sein:
- Livider Hautausschlag,
- Rezidivierende Sinusitiden,
- Kurzatmigkeit,
- Nasenbluten,
- Bluthusten aufgrund von Alveolarblutungen.
Das Churg-Strauss-Syndrom betrifft vor allem:
- Lunge,
- Herz,
- Peripheres Nervensystem.
Der Ausbruch erfolgt bei asthmatischen Krisen und peripherer Hypereosinophilie.
Nach einer variablen Periode von Monaten oder Jahren beginnt die vaskulitische Phase mit Symptomen von:
- Perikarditis,
- Myokarditis,
- Lungenentzündung,
- Mononeuritis,
- Störungen anderer Organe oder Organsysteme.
Ein positiver ANCA wird bei etwa 40 % der Patienten beobachtet.
Das Churg-Strauss-Syndrom, die Wegener-Granulomatose und die mikroskopische Polyarteriitis sind durch das Vorhandensein von ANCA im Blutserum gekennzeichnet, während Ablagerungen von Immunglobulinen an den Gefäßwänden mehr oder weniger fehlen.
Die Purpura Schönlein-Henoch und die gemischte essentielle Kryoglobulinämie sind durch Immunkomplexe in den Gefäßwänden gekennzeichnet, während die Suche nach ANCA negativ ist.
Kutane Vaskulitis
Die kutane Vaskulitis kann eine wichtige Komponente der systemischen Vaskulitis sein, wie:
- Die der rheumatoiden,
- Die der mit ANCA assoziierten (Wegener-Granulomatose, Churg-Strauss-Syndrom, mikroskopische Polyangiitis).
Das charakteristische Zeichen ist eine tastbare Purpura, die eine oberflächliche Vaskulitis in den kleinen Gefäßen aufweist.
Weniger oft entwickeln sich:
- Ein knotiges Erythem,
- Livedo racemosa (rote netzförmige Flecken auf der Haut),
- Tiefe Geschwüre,
- Digitale Gangrän.
In diesen Fällen ist die Vaskulitis tiefliegend, in Höhe der Lederhaut oder des Unterhautgewebes.
Zur Stellung der Diagnose ist eine Biopsie erforderlich, die bis in das Unterhautgewebe reicht, entnommen an der schmerzhaftesten, am stärksten geröteten oder bläulich verfärbten Stelle.
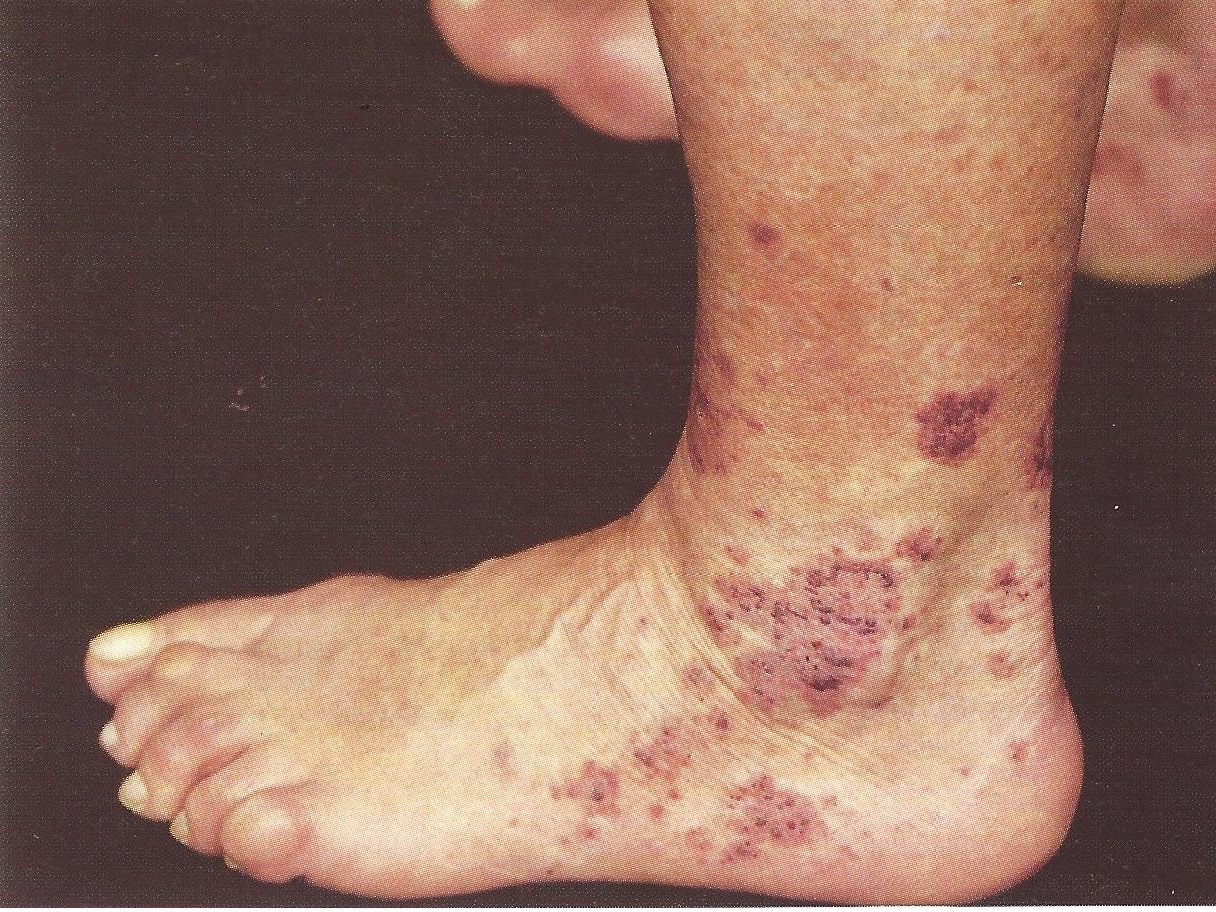
Bei den kutanen Vaskulitiden ist die häufigste Form die leukozytoklastische (oder hypersensitive) Vaskulitis, die vor allem verursacht:
- Purpura,
- Lokale Nekrosen an Füßen und Knöcheln,
- Blasen,
- Papeln,
- Knoten,
- Netzartige, blau-violette Zeichnungen.
Sie kann auch Ulzerationen hervorrufen.
Die Urtikaria-Vaskulitis ist eine Form der Vaskulitis, die die Haut befällt, indem sie verursacht:
- Quaddeln oder Nesselausschlag,
- Rote Flecken.
Es gibt zwei Formen:
- Eine mit normaler Konzentration von Komplement-Proteinen im Blut (normokomplementämisch).
- Die andere mit geringen Mengen an Komplement-Proteinen, genannt Vaskulitis mit Hypokomplementämie.
Die Urtikariavaskulitis ist eine Überempfindlichkeitsreaktion vom Typ III, bei der die Antigen-Antikörper-Komplexe in der Gefäßschicht abgelagert werden.
Diese Reaktion verursacht:
- Aktivierung des Komplements,
- Anziehung von Neutrophilen.
Diese Zellen setzen verschiedene proteolytische Enzyme frei, zum Beispiel Kollagenase und Elastase. Diese verursachen Schäden in der Gefäßwand.
Die häufigsten Symptome sind:
- Juckreiz,
- Schmerzen,
- Brennendes Gefühl.
Die Flecken auf der Haut haben oftmals einen roten Rand mit einem weißen Zentrum.
Unter der Haut können sich befinden:
- Petechien,
- Einblutungen.
In den meisten Fällen ist diese Erkrankung idiopathisch oder hat keine spezifischen Ursachen. Ärzte verbinden sie jedoch mit:
- Manchen Störungen des Immunsystems,
- Leukämie,
- Sjögren-Syndrom,
- Systemischem Lupus.
Außerdem können manche Viruserkrankungen eine Urtikariavaskulitis verursachen, wie:
- HBV (Hepatitis B),
- HCV (Hepatitis C),
- Herpes-Zoster-Virus,
- Infektiöse Mononukleose.
Die Urtikariavaskulitis könnte die Ursache des Lupus sein.
Die Krankheit könnte auch eine Nebenwirkung auf Medikamente sein, wie:
- Penicillin,
- ACE-Hemmer,
- Antidepressiva.