Alpha- und Beta-Thalassämie – Minor und Major

Die Thalassämie ist eine Erbkrankheit, die durch einen Defekt des Hämoglobins verursacht wird.
Hämoglobin ist ein Pigment, das in den roten Blutkörperchen enthalten ist und sich bindet an:
- Sauerstoff
- Kohlendioxid.
Diese Bindung ermöglicht den roten Blutkörperchen den Transport von Sauerstoff zu den Zellen und von Kohlendioxid zu den Lungen.
Das Hämoglobin enthält 2 Alpha- und 2 Betaketten, die miteinander verbunden sind.
Es ist für die richtige Funktion und das Überleben dieser Zellen wichtig, dass die Anzahl der Alpha- und Beta-Globinketten in den roten Blutkörperchen (Erythrozyten) gleich ist.
Das Fehlen von Alpha-Hämoglobinketten verursacht eine Anhäufung von Beta-Hämoglobin in den roten Blutkörperchen und umgekehrt.
Die Folge ist die Bildung von Präzipitaten (Kettengruppen), die die Erythrozyten schädigen können.
Diese Aggregate oxidieren und können verursachen:
- Hämolyse (Tod der roten Blutkörperchen), weil sie die Zellmembran schädigen,
- Unwirksame Erythropoese (Bildung roter Blutkörperchen).
Die Schwere der Erkrankung hängt vom Ungleichgewicht der Anzahl an Globinketten ab.
Inhalt
Verlauf und Folgen der Thalassämie
Die Abnahme der roten Blutkörperchen verursacht eine Anämie und der Körper versucht, diese zu kompensieren, indem er die Eisenabsorption aus der Nahrung erhöht.
Die Milz filtert die geschädigten Erythrozyten heraus und vernichtet sie.
Die Folgen der Thalassämie in der Milz sind:
- Überlastung,
- Splenomegalie (Organvergrößerung).
Mit der Zeit hält die vergrößerte Milz auch die weißen Blutkörperchen und die Blutplättchen zurück.
Die Folgen können sein:
- Infektionen,
- Blutungen.
Folgen der Thalassämie sind:
- Anoxie (Sauerstoffmangel in den Zellen),
- Wachstumsverzögerung bei Kindern und verzögerte Pubertätsentwicklung.
- Gallensteine, denn die übermäßige Produktion von Bilirubin aufgrund chronischer Hämolyse ist die Grundvoraussetzung zur Bildung von Pigmentgallensteinen (EVERSON G. T.: Gallbladder function in gallstone disease. Gastroenterol. Clin. North Am. 20 (1991), 85).
- Knochenveränderungen und Osteoporose aufgrund der Zunahme und Hyperaktivität des Knochenmarks (im Knocheninneren),
- Die Knochenmarksexpansion trägt dazu bei, dass sich der kortikale Teil des Knochens abnutzt, woraus eine Knochenneubildung folgt: „Bürstenschädel“.
Röntgenaufnahme – Bürstenschädel als Folge der Thalassämie.[/caption]
Notwendige Bluttransfusionen führen zu einer Anhäufung von Eisen.
Eisen wird vom Körper benötigt, doch ein Atom von freiem Eisen führt zur Bildung freier Radikale, die für den Organismus giftig sind.
Im Körper bindet sich Eisen immer an Proteine: Ferritin, Transferrin usw.
Sind diese Proteine mit Eisen gesättigt, häuft sich dieses Metall in den Organe an:
- Leber (zuerst betroffen), das kann zu chronischer Hepatitis und Zirrhose führen,
- Herz (wenn die Leber bereits gesättigt ist), das führt zu Rhythmusstörungen und Herzinsuffizienz,
- Drüsen, die Folgen können sein: Hypogonadismus, Hypothyreose, Diabetes, Hypoparathyreoidismus, Quelle: Borgna-Pignatti C – (Department of Clinical and Experimental Medicine, University of Ferrara and Division of Pediatrics, Arcispedale Sant’Anna, Via Savonarola 9, 44100, Ferrara, Italy)
Der Tod erfolgt durch einer Herzerkrankung und nicht aufgrund einer Erkrankung der Leber, weil eine kleine Menge Eisen ausreicht, die Nervenleitung des Herzens zu verändern.
Wie wird die Thalassämie übertragen?
Die Ursache der Thalassämie ist immer genetisch bedingt: sie ist eine autosomal-rezessive Erkrankung. Ein oder beide Elternteile sind gesunde Träger der Erkrankung.
Die Gene können auf ihre Kinder übergehen.
Einteilung der Thalassämie
Es gibt zwei Arten von Globinketten: Alpha und Beta.
Alpha
- Gesunder Träger
- Thalassämie-Merkmal
- HbH-Krankheit
- Hydrops fetalis – die schwerste Form der Alpha-Thalassämie, sie führt zum Tod des Fötus oder des Neugeborenen.
Beta
- Minor, in der Regel ohne Symptome,
- Intermedia,
- Major.
Alpha-Thalassämie
Die Alpha-Thalassämie verursacht einen Überschuss an Beta-Globin, was zur Bildung von Beta-Globin-Tetrameren (β4) führt. Diese Krankheit wird als Hämoglobin-H-Krankheit bezeichnet.
Die β4 Tetramere sind löslich, können aber im Falle einer Oxidation Hämolyse verursachen.
Die Hämoglobin-Constant-Spring-Krankheit ist eine schwere Form dieser hämolytischen Störung.
Die schwerste Form der Thalassämie ist die Alpha-Thalassämie major, bei der ein Fötus kein Alphaglobin produziert. Diese Situation ist mit dem Leben nicht vereinbar.
Die Gene des Alpha-Globins befinden sich auf Chromosom 16.
Die Alpha-Kette des Hämoglobins besteht aus 4 kugelförmigen Untereinheiten.
Jede Untereinheit ist durch ein anderes Gen im Chromosom kodiert.
Die Symptome und Komplikationen der Erkrankung hängen von der Zahl der fehlenden oder mutierten Gene ab:
- Ein gesunder Träger hat keine Anzeichen der Erkrankung.
- Bei zwei abnormen (oder fehlenden) Genen entsteht eine Thalassämie vom Typ Alpha oder eine Thalassämie minor. Diese Erkrankung verursacht eine leichtgradige Anämie.
- Wenn drei Gene mutiert sind oder fehlen, entwickelt der Patient eine Hämoglobin-H-Krankheit (durch Blutuntersuchung diagnostiziert). Diese Form der Thalassämie verursacht eine mittelgradige oder schwere Anämie.
- Wenn alle Gene fehlen oder eine Mutation erfahren haben, ist die Thalassämie sehr schwer ausgeprägt und das Kind wird tot geboren oder der Tod tritt kurz nach der Geburt ein.
Ein Kind erbt vier Alpha-Globine (zwei von jedem Elternteil).
Das Kind zweier von Thalassämie betroffenen Personen (beide mit zwei fehlenden oder mutierten Genen) hat:
- Eine Wahrscheinlichkeit von 25 % zu erkranken (3 oder 4 anomale Gene),
- Eine Wahrscheinlichkeit von 50 %, ein gesunder Träger zu sein,
- 25 %, gesund zu sein oder ein abnormes Gen zu besitzen.
Fehlen dem Vater zwei Alpha-Globin-Gene und der Mutter ein Alpha-Globin-Gen, hat jedes Kind die Wahrscheinlichkeit von 25 %, folgende Genkombinationen zu erben:
- Vier normale Gene (keine Anämie),
- Ein fehlendes Gen und drei normale Gene (gesunder Träger),
- Zwei „fehlende“ Gene und zwei normale Gene (Krankheit),
- Drei fehlende Gene und ein normales Gen (H-Hämoglobinopathie),
Die meisten Menschen mit Alpha-Thalassämie zeigen nur leichte Symptome der Erkrankung.
Beta-Thalassämie
Die Beta-Thalassämie verursacht einen Überschuss an Alpha-Globin, das alpha-Globin-Tetramere (α4) bildet, anstatt zwei Alphakettenpaare und zwei Betakettenpaare.

Diese unlösliche Bildung reichert sich im Erythroblasten (unreifes rotes Blutkörperchen) an und beeinträchtigt:
- Die Erythropoese,
- Die Zellreifung,
- Die Zellmembran.
Folge ist eine Anämie und die Bildung von nicht funktionierenden roten Blutkörperchen.
Die Betaketten werden von zwei verschiedenen Genen kodiert.
Im Falle des Fehlens oder einer Mutation:
- Von nur einem Gen, ist die Anämie nur leichtgradig ausgeprägt oder fehlt ganz,
- Von beiden Genen, sind die Symptome schwerwiegend.
Das Beta-Globin-Gen befindet sich auf Chromosom 11.
Ein Kind erbt zwei Beta-Globin-Gene (eines von jedem Elternteil). Fehlen diese Gene oder sind sie verändert, kommt es zur Beta-Thalassämie. Das bedeutet, dass der Organismus nicht genug Beta-Ketten des Hämoglobins produziert.
- Wer nur ein verändertes Gen besitzt, ist ein gesunder Träger, denn es handelt sich um ein rezessives Gen. Diese Erkrankung wird als Beta-Thalassämie minor bezeichnet und verursacht eine leichtgradige Anämie.
- Wenn beide Gene verändert sind oder fehlen, entsteht eine Beta-Thalassämie intermedia oder Beta-Thalassaemia major (auch Cooley-Anämie genannt).
Die intermediale Form verursacht eine milde Verlaufsform der Anämie. Die Major-Form verursacht eine schwere Anämie.
Wenn jedes Elternteil ein verändertes Gen hat, hat jedes Kind:
- Eine Wahrscheinlichkeit von 25 %, zwei normale Gene (keine Anämie) zu erben,
- Eine Wahrscheinlichkeit von 50 %, ein verändertes und ein normales Gen zu erben (Beta-Thalassämie minor), in diesem Fall kann der Patient eine leichte Anämie haben oder er hat im Falle einer minimalen Thalassämie keinerlei Symptome.
- Eine Wahrscheinlichkeit von 25 %, zwei veränderte Gene (beta-Thalassämie major) zu erben.
Unterschied zwischen Thalassämie major und minor
Es gibt zwei Formen der Beta-Thalassämie: die Thalassämie minor (oder gutartige Anämie) und die Thalassämie major (bekannt als Cooley-Anämie).
Thalassämie minor: der Betreffende hat ein verändertes oder fehlendes Gen (zusammen mit dem Gen einer normalen Beta-Kette). Er wird als heterozygoter Träger einer Beta-Thalassämie bezeichnet.
Menschen mit einer Thalassämie minor haben eine leichtgradige Anämie mit geringer Absenkung des Hämoglobins im Blut.
Menschen mit Thalassaemia minor haben einen normalen Eisenspiegel im Blut. Es ist keinerlei Behandlung der Thalassaemia minor erforderlich.
Die Thalassaemia major (oder Cooley-Anämie): Die Betroffenen mit einer Thalassaemia major besitzen zwei veränderte Gene einer „Beta-Thalassämie“ und kein normales Beta-Gen.
Der Betroffene ist homozygoter Träger einer Beta-Thalassämie.
Das verursacht einen Mangel in der Betaketten-Produktion.
Die Thalassaemia major ist eine schwerwiegende Krankheit.
Das klinische Bild der Thalassaemia major wurde 1925 von dem amerikanischen Kinderarzt Thomas Cooley beschrieben.
Ein Kind mit einer Thalassaemia major erscheint bei seiner Geburt gesund.
Das bei der Geburt vorherrschende Hämoglobin ist noch immer das fetale Hämoglobin (HbF). Das HbF enthält zwei Alpha-Ketten (wie das adulte Hämoglobin HbA) und zwei Gamma-Ketten (im Gegensatz zum HbA).
Da das Kind keine Beta-Kette hat, ist es bei seiner Geburt von den Effekten der Thalassaemia major geschützt.
Favismus ist eine durch einen Enzymmangel verursachte Pathologie: Mangel an Glucose-6-Phosphat-Dehydrogenase oder G-6-PDH.
Menschen, die unter dieser Krankheit leiden, können wegen der Zerstörung der roten Blutkörperchen eine Anämie mit Ikterus erleiden, wenn sie Bohnen, Erbsen und andere Gemüsearten essen.
Das kann auch nach Einnahme mancher Medikamente wie Salicylate und Sulfonamide auftreten.
Thalassämie in der Schwangerschaft
Generell ist eine Schwangerschaft bei Frauen mit einer β-Thalassämie möglich und sicher.
Heute können auch Frauen mit einer Thalassämie major oder Thalassämie intermedia Kinder haben. (Thalassaemia in pregnancy – Leung TY – Lao TT Best Pract Res Clin Obstet Gynaecol. 2012 Feb; 26 (1): 37-51).
Die wichtigsten zu bewertenden Faktoren sind Messungen der Herzfunktion und der Eisenbelastung mittels einer Magnetresonanztomographie.
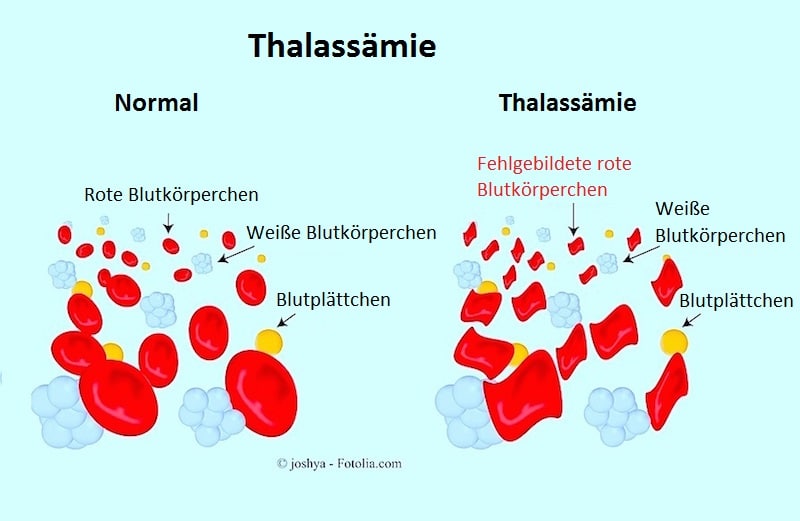
Symptome der Thalassämie
Die Symptome der Beta-Thalassämie hängen von der Schwere der Erkrankung ab.
Gesunde Träger zeigen keinerlei Symptome.
Die von einer schwerwiegenden Thalassämie Betroffenen können ab den ersten Monaten ihres Lebens folgende Symptome aufweisen:
- Hochgradige Anämie,
- Müdigkeit und Schwäche,
- Kurzatmigkeit,
- Vermindertes Wachstum,
- Gesichtsblässe (eines der ersten Symptome, das bei Neugeborenen beobachtet wird),
- Dunkler Harn,
- Vergrößerung der Leber und der Milz,
- Ikterus.
Wer Sport treibt und einen Hämoglobinwert unter 9 g/dl hat, kann einen deutlichen Leistungsabfall aufweisen.
Zu den Konsequenzen der Mittelmeeranämie zählt das Verbot zur Blutspende, wenn die Hämoglobinwerte niedriger sind als:
- 13,5 g/dl bei Männern,
- 12,5 g/dl bei Frauen.
Diagnose der Mittelmeeranämie
Der Arzt diagnostiziert eine leichte und schwere Thalassämie in der Kindheit, weil die Symptome bereits in den ersten beiden Lebensjahren auftreten.
Man benutzt verschiedene Blutuntersuchungen, um die Thalassämie zu diagnostizieren:
Ein komplettes Blutbild umfasst:
- Hämoglobinwerte,
- Anzahl und Größe der roten Blutkörperchen.
Menschen mit Thalassämie haben weniger rote Blutkörperchen und weniger Hämoglobin als normal.
Menschen mit Alpha-/Beta-Thalassämie können kleinere rote Blutkörperchen haben.
Ein Erwachsener sollte höhere Hämoglobin- und Hämatokritwerte haben als:
- Erwachsener Mann – 13 g/dl Hämoglobin und 39 % Hämatokrit;
- Erwachsene Frau – 12 g/dl Hämoglobin und 36 % Hämatokrit;
- Schwangere Frau – 11 g/dl Hämoglobin und 33 % Hämatokrit.
Die Anzahl der Retikulozyten (Messung der Zellen, aus denen die Erythrozyten entstehen) kann einen Hinweis darauf geben, ob das Knochenmark eine ausreichende Anzahl roter Blutkörperchen produziert.
Laboruntersuchungen des Eisenspiegels weisen darauf hin, ob die Ursache für die Anämie ist:
- Eisenmangel,
- Thalassämie.
Man darf die Ferritinwerte nicht mit dem Eisengehalt im Blut verwechseln, denn das sind zwei verschiedene Daten.
Bei Betroffenen der Mittelmeeranämie kann das Eisen normal sein.
Das indirekte Bilirubin (unkonjugiert) ist bei Patienten mit Thalassämie höher.
Besteht eine familiäre Krankengeschichte mit Thalassämie, wird ein genetischer Test zur Diagnostik hinzugezogen.
Um eine pränatale Mittelmeeranämie zu diagnostizieren, muss eine Chorionzottenbiopsie des Fötus bereits ab der 10. Woche erfolgen. Auf diesem Weg kann man erfahren, ob das Kind an einer Thalassämie erkranken wird.
Wie wird eine Thalassämie behandelt?
Die Standardtherapie besteht in:
- Bluttransfusion,
- Eisenausschleusender Therapie (Chelation).
In besonders schweren Fällen kann man durchführen:
- Entfernung der Milz,
- Knochenmarktransplantation.
Die Bluttransfusion umfasst die Übertragung von Blut in die Vene, um normale Werte von roten Blutkörperchen und Hämoglobin wiederherzustellen.
Die Transfusionen werden wiederholt:
- Alle 4 Monate bei mittelgradiger Thalassämie,
- Alle 2-4 Wochen bei einer Beta-Thalassämie.
Bei der Eisenchelation erfolgt die Entfernung überschüssigen Eisens aus dem Körper.
Bluttransfusionen können eine Überladung mit Eisen verursachen.
Patienten, die Bluttransfusionen erhalten, müssen sich der Therapie einer Eisenchelation unterziehen.
Die verwendeten Pharmaka bei der Eisenchelation sind:
- Deferoxamin, eine Lösung zur subkutanen Verabreichung,
- Deferasirox, eine Tablette, die oral eingenommen wird.
Die Splenektomie (Entfernung der Milz) kann bei Patienten mit einer Hämoglobin-H-Krankheit erforderlich sein.
Die Knochenmarktransplantation ist die wirksamste Behandlungsmethode. Die Kompatibilität zwischen Spender und Empfänger besteht, wenn der Spender die gleichen Arten von Proteinen (humane Leukozyten-Antigene HLA) wie die Empfängerzellen auf der Oberfläche hat.
Die Transplantation des Knochenmarks unter Geschwistern ist die beste Behandlungsmöglichkeit.
Den meisten Patienten mit Thalassämie fehlt ein geeigneter Spender.
Die Stammzellen des transplantierten Knochenmarks beginnen, neue Blutzellen zu erzeugen.
Ernährung und Diät bei Thalassämie
Bei einer Thalassämie major sollten Nahrungsmittel mit hohem Eisengehalt eingeschränkt werden.
Eine verstärkte Absorption von Eisen im Darm ist für die Thalassämie charakteristisch.
Eine Tasse Schwarztee während der Mahlzeit verringert die Eisenaufnahme aus den Speisen, besonders bei der Thalassämie intermedia (Alarcon, 1979).
Es gibt jedoch keinen Nachweis, dass eine eisenarme Diät bei der Thalassämie major hilfreich wäre; es sollten nur Speisen vermieden werden, die viel Eisen enthalten, wie:
- Leber,
- Schokolade,
- Hülsenfrüchte,
- Einige Vitamincocktails.
Man sollte auf Lebensmittel für Kinder, wie Frühstücksflocken und Multivitaminprodukte, achten, da sie viel Eisen enthalten.
Calcium
Viele Thalassämie-Faktoren verursachen einen Calciummangel. Daher wird eine Ernährung empfohlen, die reich an diesem Mineral ist.
Um eine Steinbildung zu vermeiden, sollte auf Nahrungsergänzungsmittel verzichtet werden.
Gemäß der Blutgruppendiät hat sich die Thalassämie vor allem in den Mittelmeerländern entwickelt, weil in diesen Ländern die Bewohner stets viel Gluten verzehrt haben.
Nach dieser Theorie kann in den meisten Fällen eine Vermeidung von glutenhaltigem Getreide die Symptome verringern. Deshalb sollten vermieden werden:
- Nudeln,
- Cracker,
- Knabbergebäck,
- Pizza,
- Gebäck,
- Dinkel,
- Gerste,
- Hafer,
- Kamut,
- Sorghum.
Wie ist die Prognose für Thalassämiepatienten?
Patienten mit einer leichtgradigen Thalassämie haben eine gute Überlebenschance, wenn sie sich der notwendigen Behandlung unterziehen (Transfusionen und Eisenchelatbehandlung).
Die Eisenüberladung ist die hauptsächliche Todesursache bei Thalassämiepatienten, die Eisenchelattherapie ist daher ausgesprochen wichtig.
Die Transplantation von Knochenmark kann die Thalassämie definitiv heilen.
Vorbeugung der Thalassämie
Gesunde Träger einer Beta-Thalassämie müssen über die damit verbundenen Risiken bezüglich der Fortpflanzung mit einem Partner mit gleichen genetischen Veränderungen informiert werden.