Retinoblastom des Auges
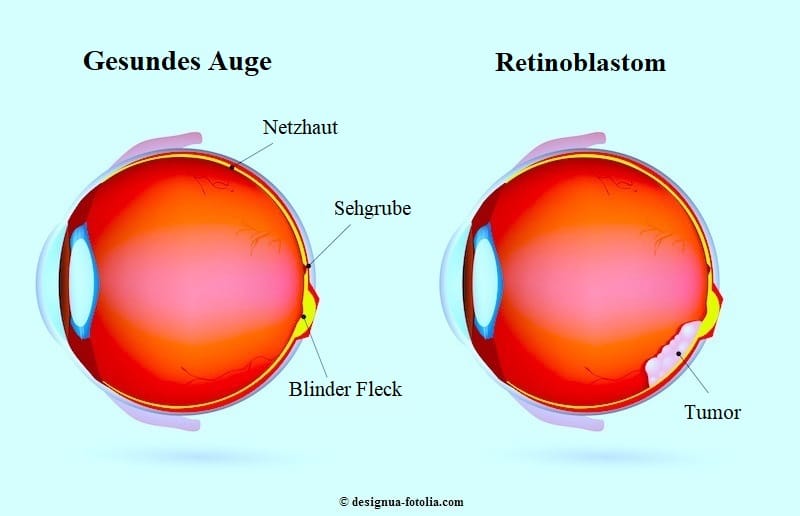
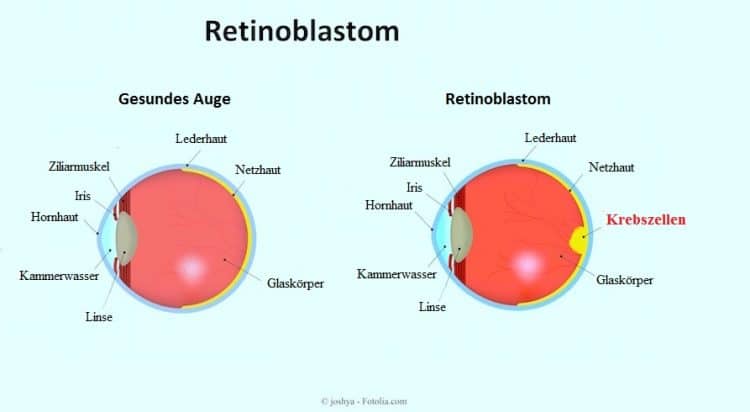
Das Retinoblastom ist ein bösartiger Tumor, der die Netzhaut im hinteren Bereich des Auges betrifft.
Es ist die häufigste Krebsart des Auges bei Kindern.
In der Regel sind Kinder unterhalb von 5 Jahren davon betroffen, sehr selten kommt es bei Erwachsenen vor.
Bei den meisten Kindern mit Retinoblastom betrifft die Krankheit nur ein Auge. Jedoch entwickelt eins von drei Kindern den Krebs in beiden Augen.
Das häufigste Anzeichen für ein Retinoblastom ist ein sichtbarer Reflex in der Pupille, auch Leukokorie (Katzenaugen-Reflex) genannt.
Dieser ungewöhnliche, weiße Reflex wird auf Fotografien, die mit Blitzlicht gemacht wurden, besonders deutlich.
 Häufigkeit
Häufigkeit
Jährlich entwickelt etwa 1 von 20.000 neugeborenen Kindern in den Vereinigten Staaten ein Retinoblastom.
Das Retinoblastom macht etwa 3 Prozent aller bösartigen Tumoren in der Kindheit aus.
Inhalt
Wie entwickelt sich ein Retinoblastom?
Die Augen entwickeln sich sehr schnell, solange sich die Kinder noch im Uterus befinden:
- In den frühen Entwicklungsphasen haben die Augen Zellen, sogenannte Retinoblasten, die sich in neue Zellen teilen, um die Netzhaut auszufüllen.
- Zu einem bestimmten Zeitpunkt hören die Zellen auf, sich zu teilen und werden zu reifen Netzhautzellen.
- In seltenen Fällen wachsen einige Retinoblasten weiterhin schnell und unkontrolliert, statt die speziellen Zellen zu entwickeln, die das Licht empfangen, und bilden den als Retinoblastom bekannten Krebs.
Die Kette von Ereignissen, die zu einem Retinoblastom führt, ist komplex, beginnt aber mit einer Anomalie (Mutation oder Modifikation) eines Tumor-Supressor-Gens, dem sogenannten Retinoblastom-Eiweiß (RB oder RB1).
Das normale RB1-Gen blockiert das unkontrollierte Wachstum der Zellen, denn es wirkt auf den Zellzyklus ein.
Dieses Gen ist auf der Bande q14 des Chromosoms 13 lokalisiert (Kürzel 13q14).
Je nach der Veränderung, die in dem RB1-Gen entsteht, können sich 2 verschiedene Arten des Retinoblastoms bilden.
Hereditäres Retinoblastom
In der Regel tritt dieser Tumortyp in einem jüngeren Alter auf als das nicht hereditäre Retinoblastom.
Das beidseitige Retinoblastom ist gewöhnlich hereditär, während das einseitige nicht hereditär ist.
Genetische Mutationen, die das Risiko für ein Retinoblastom und andere Tumore erhöhen, können von den Eltern auf die Kinder übergehen.
Das hereditäre Retinoblastom geht nach einem dominant autosomalen Muster von den Eltern auf die Kinder über:
- Das bedeutet, dass eine einzige Kopie des mutierten Gens eines Elternteils ausreichend ist, um das Risiko bei den Kindern zu erhöhen.
- Hat ein Elternteil ein mutiertes Gen, hat jedes Kind zu 50 % die Wahrscheinlichkeit, dieses Gen zu erben.
Sporadisches Retinoblastom (nicht hereditär)
Bei den meisten Kindern ist das Retinoblastom nicht hereditär.
Etwa 40 % der mit diesem Defekt geborenen Kinder haben von einem Elternteil dieses Gen geerbt.
In etwa 60 % der Fälle entwickelt sich die Genvariation noch im Uterus.
Da alle Zellen des Körpers das modifizierte RB1-Gen haben, besteht bei dieser Erkrankung ein erhöhtes Risiko, andere Tumore außerhalb des Auges zu entwickeln.
Insbesondere sind folgende Entwicklungen wahrscheinlicher:
- Manche Kinder mit dieser Form des Retinoblastoms entwickeln einen Tumor an der Epiphyse der Hirnbasis (Pinealoblastom). Dieser wird auch trilaterales Retinoblastom
- Eine Art Knochentumor, genannt Osteosarkom.
- Tumore im Weichgewebe, zum Beispiel in Muskeln.
- Eine aggressive Krebsart der Haut und des Auges, das sogenannte Melanom.
Das sporadische Retinoblastom entwickelt sich in nur einem Auge. Es ist nicht bekannt, was die Mutation des RB1-Gens verursacht.
Ein Kind mit sporadischem (nicht hereditärem) Retinoblastom entwickelt den Tumor in nur einem Auge.
Diese Art von Retinoblastom tritt oftmals in einem späteren Alter auf als die hereditäre Form.
Klassifikation des Retinoblastoms
- Das intraokulare Retinoblastom wird in einem oder beiden Augen vorgefunden, es erstreckt sich jedoch nicht über die Augen hinaus.
- Bei einem extraokularen Retinoblastom hat sich der Krebs über das Auge hinaus auf das umgebende Gewebe oder auf andere Körperteile ausgebreitet, zum Beispiel auf Gehirn, Rückenmark, Knochenmark oder Lymphknoten.
Stadien des Retinoblastoms nach der International Classification of Retinoblastoma (ICRB)
Das intraokulare Retinoblastom wird in vier Stadien eingeteilt:
- Phase I: Der Tumor ist auf die Netzhaut begrenzt.
- Phase II: Der Tumor ist auf den Augapfel begrenzt.
- Phase III: Der Tumor breitet sich in Bereiche um das Auge aus.
- Phase IV: Der Tumor breitet sich über den Sehnerv bis zum Gehirn aus oder hat über den Blutkreislauf Metastasen in Weichgewebe, Knochen oder Lymphknoten gebildet.
Wie kann man es erkennen? Zeichen und Symptome des Retinoblastoms
Das erste Anzeichen eines Retinoblastoms ist eine weiße Pupille, die bei vollem Licht silbrig oder gelb erscheinen kann.Diese Bildung heißt Leukokorie.
Andere Symptome sind:
- Schielen (Strabismus),
- Gerötete, oftmals schmerzende Pupille,
- Pupille und Augapfel, die größer als normal erscheinen,
- andersfarbige Regenbogenhaut,
- Verminderung oder Verlust des Sehvermögens bis zur Erblindung.
Diagnose des Retinoblastoms
Die frühzeitige Diagnose des Retinoblastoms kann bedeuten, dass das Kind eine weniger intensive Therapie benötigt.
Der Augenarzt untersucht das Auge unter Anästhesie. Das Retinoblastom kann nur durch die klinische Untersuchung des Auges diagnostiziert werden, gewöhnlich wird keine Biopsie durchgeführt.
Es gibt Untersuchungen, um Position und Größe des Tumors festzustellen. Außerdem muss geprüft werden, ob eine Ausbreitung in die umgebenden Strukturen begonnen hat.
Mögliche Untersuchungen sind:
- Echographie. Dies ist eine schmerzlose Untersuchung, bei der Schallwellen zur Untersuchung des Auges und der umgebenden Strukturen benutzt werden.
- Magnetresonanztomographie. Diese Untersuchung benutzt Magnetfelder zur Erstellung eines detaillierten Bildes von Auge und Kopf.
- Der Arzt führt eine feine Nadel zwischen den Knochen der unteren Wirbelsäule ein, um eine Probe der Hirnflüssigkeit zu entnehmen. Die Flüssigkeit wird mikroskopisch untersucht um festzustellen, ob Tumorzellen darin vorhanden sind.
- Biopsie aus dem Knochenmark. Sie erfolgt um festzustellen, ob sich Tumorzellen im Knochenmark befinden.
- Knochenszintigraphie. Mit einer Serie von Röntgenaufnahmen wird geprüft, ob Anzeichen für eine Ausbreitung des Krebses auf die Knochen vorliegen.
- Blutuntersuchung. Diese Untersuchung erfolgt, um den genetischen Test des RB-Gens vorzunehmen. Die Ergebnisse aus dieser Untersuchung können erst Monate später vorliegen.
- Augenuntersuchung mit erweiterter Pupille.
- CT.
Bei meinem Sohn wurde ein Retinoblastom diagnostiziert. Sollte ich auch meine anderen Kinder untersuchen lassen?
Manche Fälle können erblich sein, daher sollte bei allen Geschwistern des betroffenen Kindes eine Untersuchung durchgeführt werden, um diese Krankheit bei ihnen auszuschließen.
Wie groß ist die Wahrscheinlichkeit, dass das nächste Kind dieselbe Erkrankung hat?
Die Möglichkeiten können von 1 von 15.000 bis zu 45 % variieren und hängen von vielen Faktoren ab, darunter der familiären Disposition des Retinoblastoms, wenn der Tumor beide Augen betrifft usw.
Link zur Retinoblastom-Seite der Deutschen Krebsgesellschaft e.V.: https://www.krebsgesellschaft.de/onko-internetportal/basis-informationen-krebs/krebsarten/weitere-krebsarten/retinoblastom/ursache-und-risikofaktoren.html
Therapie des Retinoblastoms
Augenarzt und Onkologe arbeiten bei der Behandlung eines Kindes mit Retinoblastom zusammen.
Dieses Team stellt für jeden Patienten einen individuellen Behandlungsplan zusammen, je nach Ausbreitung der Erkrankung im Augeninneren, ob ein oder beide Augen betroffen sind und ob eine Verbreitung über das Auge hinaus besteht.
Es gibt viele Behandlungsarten beim Retinoblastom, alle haben das Ziel, die Tumorzellen abzutöten.
Folgende Behandlungen können verordnet werden:
- Systemische Chemotherapie: Medikamente, die den Tumor töten, werden oral oder intravenös durch eine Injektion verabreicht.
- Intraarterielle Chemotherapie: In manchen Fällen injizieren Ärzte die Chemotherapeutika direkt in die Blutgefäße, die das Auge versorgen, damit sie direkt auf den Tumor einwirken können.
- Strahlentherapie mit externen Strahlen: die Strahlen werden genau auf den Tumor fokussiert, um die Tumorzellen abzutöten.
- Brachytherapie: Hierbei handelt es sich um eine Art interne Strahlentherapie, bei der das radioaktive Material (Kapsel) im Innern des Tumors platziert wird, um die Strahlung auf bestimmte Bereiche zu konzentrieren. Diese Behandlungsform minimiert die Schäden auf das gesunde umgebende Gewebe.
- Strahlenträger: Eine Scheibe mit einer enthaltenen Strahlendosis wird direkt auf den Tumor aufgelegt.
- Kryotherapie: Flüssiger Stickstoff oder Argongas, zwei extrem kalte Substanzen, werden verwendet, um das erkrankte Gewebe einzufrieren und zu zerstören.
- Transpupillare Thermotherapie: Laserenergie erwärmt die Tumorzellen und die umgebenden Blutgefäße, um die Krebszellen abzutöten.
- Photokoagulation: Laserenergie wird auf Blutgefäße geleitet und führt zur Bildung von Blutgerinnseln, durch die den Tumorzellen Nährstoffe entzogen werden.
- Enukleation: In schweren Fällen eines Retinoblastoms (sehr selten) wird der gesamte Augapfel entfernt, um eine Verbreitung der Krebszellen auf andere Bereiche des Körpers zu verhindern. Anstelle des entfernten Augapfels kann eine Augenprothese (Glasauge) eingesetzt werden.
Wie sind die Lebenserwartungen bei einem Retinoblastom?
Obwohl das Retinoblastom eine gefährliche Erkrankung ist, ist es selten tödlich, wenn es entsprechend behandelt wird.
Mit der richtigen und frühzeitig durchgeführten Behandlung durch einen erfahrenen Augenarzt liegt die Heilungsrate der Patienten bei 95 %.
Die Prognose hängt von folgenden Faktoren ab:
- Schwere der Erkrankung.
- Größe und Sitz des Tumors.
- Vorhandensein oder Abwesenheit von Metastasen.
- Reaktion des Tumors auf die Therapie.
- Alter und allgemeine Gesundheit des Kindes.
- Empfindlichkeit des Kindes auf spezielle Medikamente, Verfahren oder Therapien.